Tutorial 2: 10x Visium human dorsolateral prefrontal cortex (DLPFC) datasets
1 Import modules
[1]:
import pandas as pd
import scanpy as sc
import Castl
2 Load data and gene lists
[2]:
adata = sc.read_visium(path='../data/DLPFC/151673', count_file='filtered_feature_bc_matrix.h5')
adata.var_names_make_unique()
adata.obs["x_pixel"] = pd.Series(adata.obsm['spatial'][:, 1], index=adata.obs.index)
adata.obs["y_pixel"] = pd.Series(adata.obsm['spatial'][:, 0], index=adata.obs.index)
sc.pp.log1p(adata)
D:\Apps\Anaconda3\Lib\site-packages\anndata\_core\anndata.py:1756: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
D:\Apps\Anaconda3\Lib\site-packages\anndata\_core\anndata.py:1756: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
[3]:
methods = ['spatialde', 'spark', 'sparkx', 'somde', 'spagcn', 'spanve', 'heartsvg']
gene_lists = {}
for method in methods:
file_path = f'../results/DLPFC/151673/DLPFC_151673_{method}_results_processed.csv'
var_name = f'{method}_genelist'
gene_lists[var_name] = pd.read_csv(file_path)
locals().update(gene_lists)
[4]:
methods = ['spatialde', 'spark', 'sparkx', 'somde', 'spagcn', 'spanve', 'heartsvg']
gene_lists = {}
for method in methods:
file_path = f'../results/DLPFC/151673/151673_stabl/DLPFC_151673_{method}_stabl_processed.csv'
var_name = f'{method}_combined_genelist'
gene_lists[var_name] = pd.read_csv(file_path)
locals().update(gene_lists)
3 Run Castl
3.1 Rank aggregation
[5]:
rank_agg_genelist = Castl.rank_agg(
gene_list=[spatialde_genelist, spark_genelist, sparkx_genelist,
somde_genelist, spagcn_genelist, spanve_genelist, heartsvg_genelist],
gene_col='gene',
rank_col='adjusted_p_value',
ascending=True,
top_percent=0.1
)
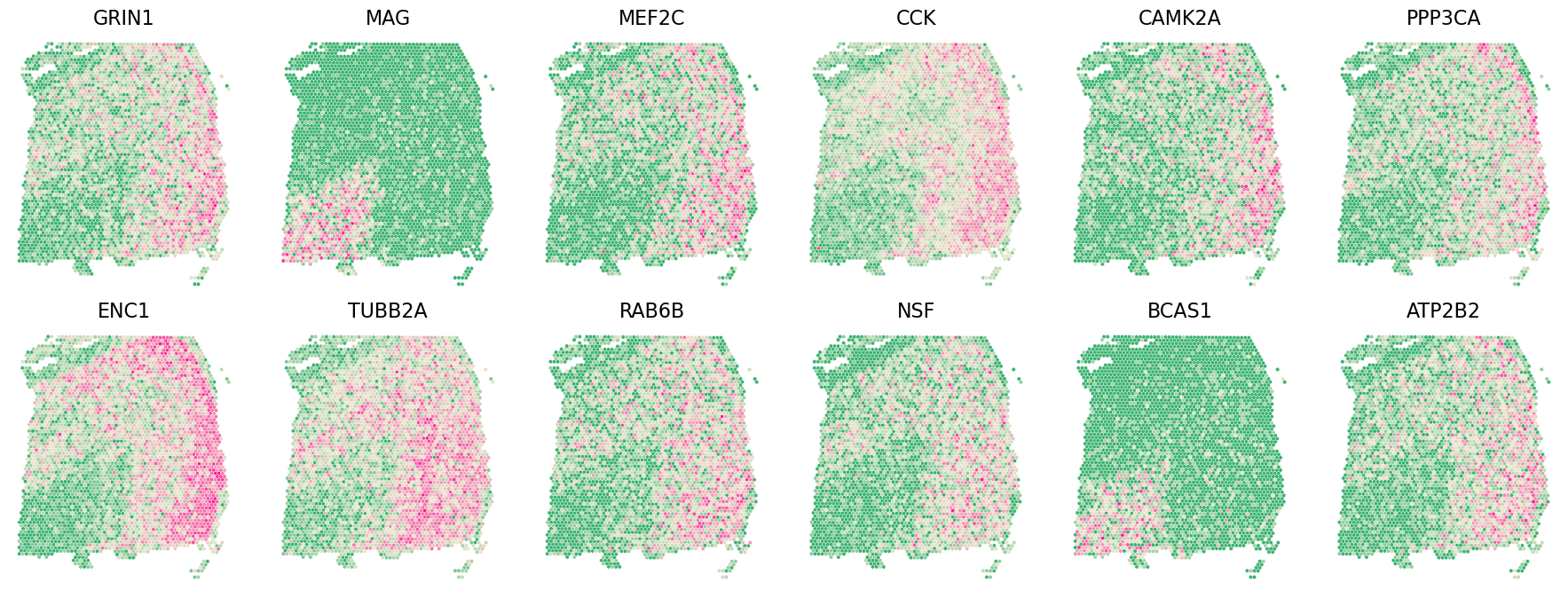
Castl.plot_gene(
adata=adata,
gene_df=rank_agg_genelist,
x_col='y_pixel',
y_col='x_pixel',
gene_col='gene',
sort_col='score',
ascending=False,
top_n=12,
spotsize=10,
figsize=(18, 6.8),
invert_yaxis=True
)
rank_agg_genelist.head()
[5]:
| gene | score | rank | pred | |
|---|---|---|---|---|
| 16201 | GRIN1 | 112491.0 | 1 | 1 |
| 10281 | MAG | 112368.0 | 2 | 1 |
| 4535 | MEF2C | 112362.0 | 3 | 1 |
| 16831 | CCK | 112322.0 | 4 | 1 |
| 15284 | CAMK2A | 112316.0 | 5 | 1 |
3.2 P-value aggregation
[6]:
pval_agg_genelist = Castl.pval_agg(
gene_list=[spatialde_genelist, spark_genelist, sparkx_genelist,
somde_genelist, spagcn_genelist, spanve_genelist, heartsvg_genelist],
gene_col='gene',
pvalue_col='adjusted_p_value',
correction='fdr_by'
)
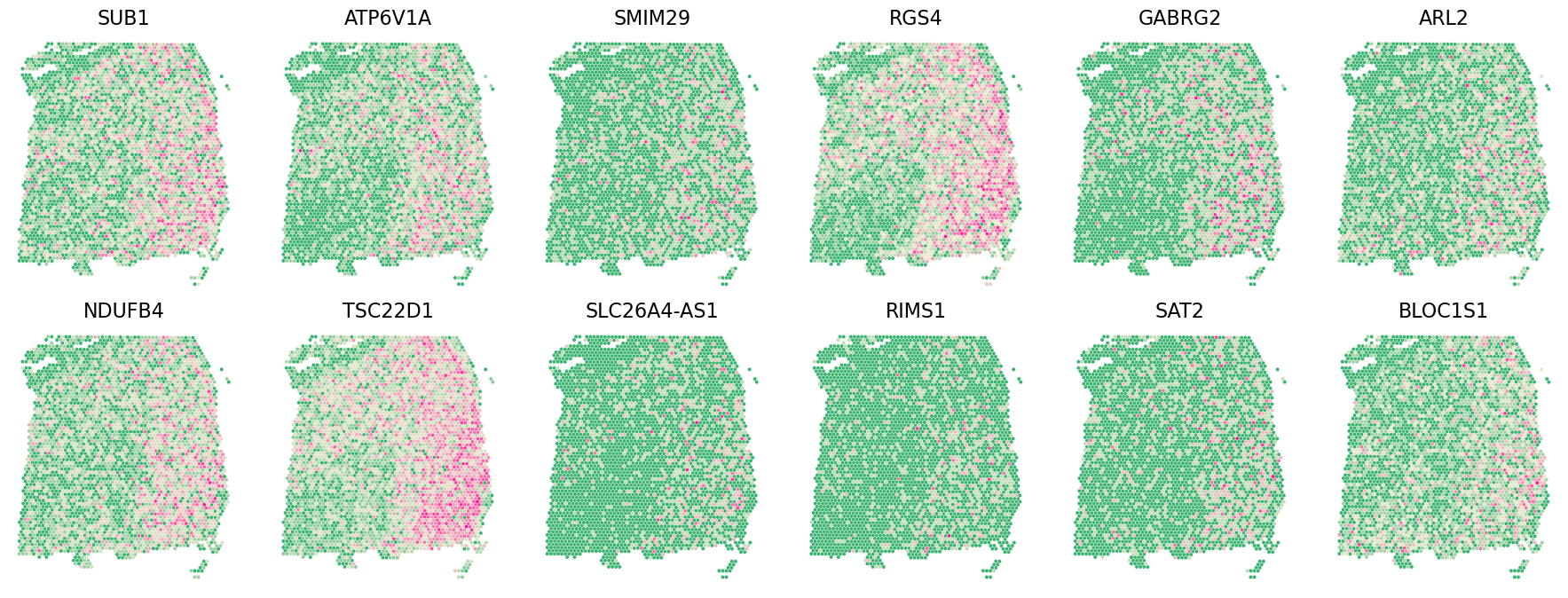
Castl.plot_gene(
adata=adata,
gene_df=pval_agg_genelist,
x_col='y_pixel',
y_col='x_pixel',
gene_col='gene',
sort_col='adjusted_p_value',
ascending=True,
top_n=12,
spotsize=10,
figsize=(18, 6.8),
invert_yaxis=True
)
pval_agg_genelist.head()
[6]:
| gene | combined_p_value | adjusted_p_value | rank | pred | |
|---|---|---|---|---|---|
| 3882 | SUB1 | 0.0 | 0.0 | 1 | 1 |
| 16812 | UQCRH | 0.0 | 0.0 | 2 | 1 |
| 16813 | ATP1A3 | 0.0 | 0.0 | 3 | 1 |
| 8815 | RPS21 | 0.0 | 0.0 | 4 | 1 |
| 16815 | MPRIP | 0.0 | 0.0 | 5 | 1 |
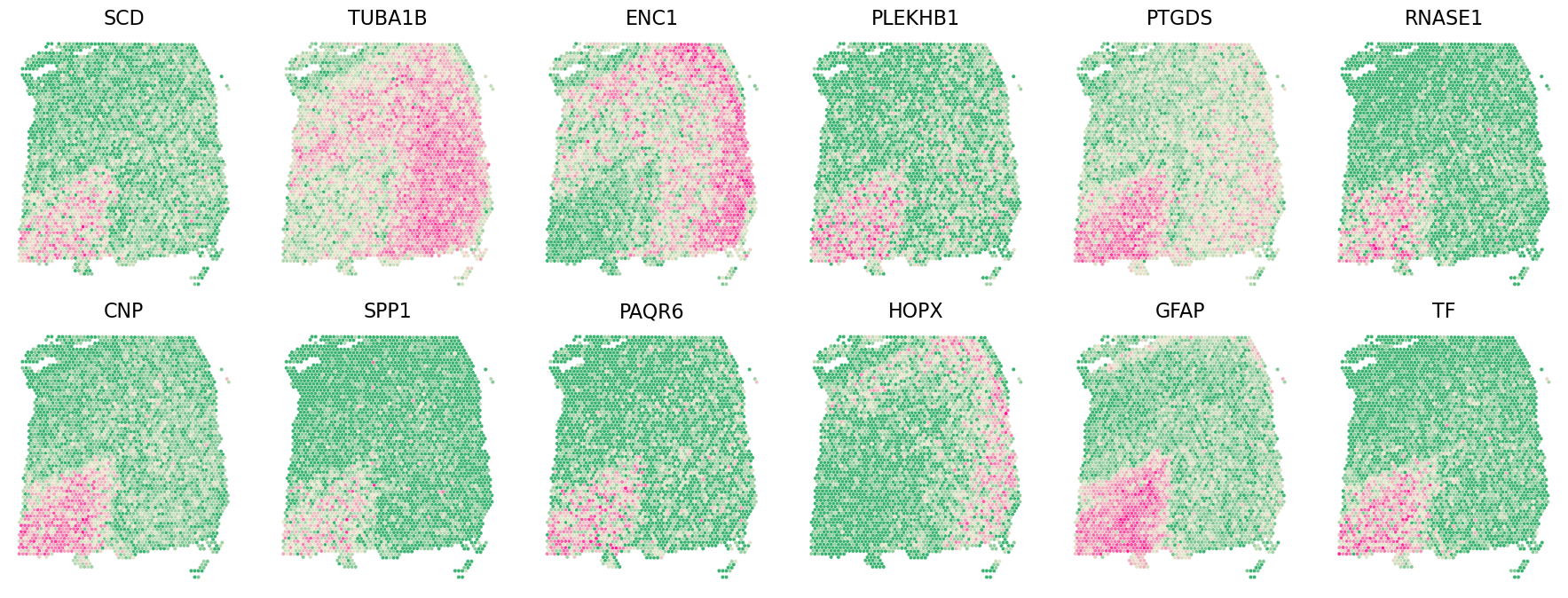
3.3 Stabl aggregation
[7]:
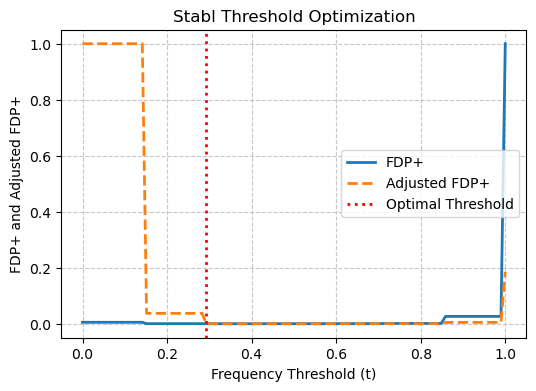
stabl_agg_genelist = Castl.stabl_agg(
gene_list=[spatialde_combined_genelist, spark_combined_genelist, sparkx_combined_genelist,
somde_combined_genelist, spagcn_combined_genelist, spanve_combined_genelist, heartsvg_combined_genelist],
gene_col = 'gene',
pred_col = 'pred',
penalty_factor=0.1,
plot=True
)
Castl.plot_gene(
adata=adata,
gene_df=stabl_agg_genelist,
x_col='y_pixel',
y_col='x_pixel',
gene_col='gene',
sort_col='frequency',
ascending=False,
top_n=12,
spotsize=10,
figsize=(18, 6.8),
invert_yaxis=True
)
stabl_agg_genelist.head()
Optimal threshold: 0.293
Total SVGs selected: 4073
ArtGene count in selection: 0
Non-ArtGene count in selection: 4073
[7]:
| gene | frequency | rank | pred | |
|---|---|---|---|---|
| 32527 | SCD | 1.0 | 1 | 1 |
| 15231 | BCAS1 | 1.0 | 2 | 1 |
| 49339 | CARNS1 | 1.0 | 3 | 1 |
| 43597 | APOD | 1.0 | 4 | 1 |
| 7431 | PLP1 | 1.0 | 5 | 1 |
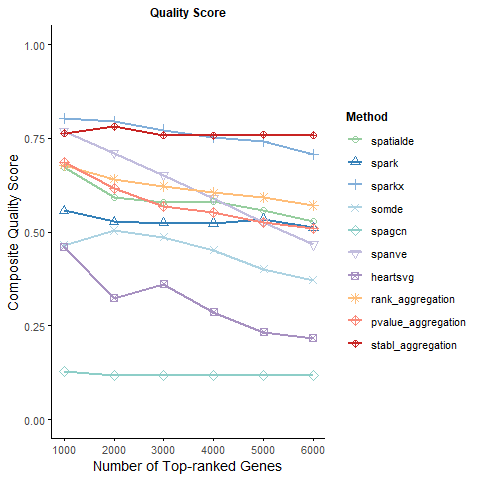
4 Calculate Quality Score
The R package clusterProfiler was employed to calculate the QS. Below is the complete procedure for executing R code within a Python kernel to compute and plot QS:
[8]:
%load_ext rpy2.ipython
D:\Apps\Anaconda3\Lib\site-packages\rpy2\robjects\packages.py:367: UserWarning: The symbol 'quartz' is not in this R namespace/package.
warnings.warn(
[9]:
%%R
library(castlRUtils)
methods <- c("spatialde", "spark", "sparkx", "somde", "spagcn",
"spanve", "heartsvg", "rank_aggregation",
"pvalue_aggregation", "stabl_aggregation")
sample_ids <- c("151507", "151508", "151509", "151510",
"151669", "151670", "151671", "151672",
"151673", "151674", "151675", "151676")
combined_df <- load_data(sample_ids, methods)
colors <- c("#97ce9f", "#3480b8", "#82afda", "#add3e2", "#8dcec8",
"#c2bdde", "#a791c1", "#ffbe7a", "#fa8878", "#c82423")
labels <- c('SpatialDE', 'SPARK', 'SPARK-X', 'SOMDE', 'SpaGCN',
'Spanve', 'HeartSVG', 'Rank Aggregation', 'P-value Aggregation', 'Stabl Aggregation')
results <- calculate_qs(
gene_list = combined_df,
sample_ids = sample_ids,
colors = colors,
method_labels = labels
)
print(results$key_metrics)
plot(results$plots$quality)
R[write to console]:
R[write to console]:
R[write to console]: castlRUtils package loaded.
Required packages are now available:
- Core packages: dplyr, ggplot2
- Analysis packages: clusterProfiler, org.Hs.eg.db, patchwork, TissueEnrich, SummarizedExperiment
All dependencies are ready to use.
# A tibble: 10 x 19
method consistency_1000 consistency_2000 consistency_3000 consistency_4000
<chr> <dbl> <dbl> <dbl> <dbl>
1 SpatialDE 0.904 0.810 0.774 0.771
2 SPARK 0.739 0.699 0.681 0.680
3 SPARK-X 0.999 1.00 0.992 0.982
4 SOMDE 0.777 0.715 0.650 0.601
5 SpaGCN 0.164 0.149 0.149 0.149
6 Spanve 0.937 0.889 0.823 0.753
7 HeartSVG 0.748 0.620 0.650 0.576
8 Rank Agg~ 0.899 0.858 0.836 0.809
9 P-value ~ 0.888 0.813 0.755 0.732
10 Stabl Ag~ 0.998 0.997 0.982 0.986
# i 14 more variables: consistency_5000 <dbl>, consistency_6000 <dbl>,
# functional_specificity_1000 <dbl>, functional_specificity_2000 <dbl>,
# functional_specificity_3000 <dbl>, functional_specificity_4000 <dbl>,
# functional_specificity_5000 <dbl>, functional_specificity_6000 <dbl>,
# quality_score_1000 <dbl>, quality_score_2000 <dbl>,
# quality_score_3000 <dbl>, quality_score_4000 <dbl>,
# quality_score_5000 <dbl>, quality_score_6000 <dbl>